具体来说,FDA 将通过一系列方法减少、改进或可能替代动物实验要求,包括基于人工智能(AI)的毒性计算模型、实验室环境中的细胞系和类器官毒性测试(即所谓的“新方法论”)。该方案将立即在新药临床试验申请(IND)中实施,鼓励纳入这些“新方法论”数据。为确定药物的有效性,FDA 还将开始使用来自其他国家的已有的真实世界安全性数据(这些国家的监管标准与美国相当,且该药物已在这些国家中进行过人体研究)。

于今年 4 月 1 日上任的 FDA 新任局长 Martin A. Makary 博士表示,长期以来,制药公司对已在国际上广泛用于人类的药物进行着额外的动物试验。此次宣布的新举措标志着药品评估方式的转变,有望在加快提供治愈方法和有效治疗的同时减少动物试验。通过利用基于人工智能(AI)的计算模型、基于人体器官模型的实验室测试以及真实世界的人类数据,我们能够更快速、更可靠地为患者提供更安全的治疗,同时降低研发成本和药品价格。这对公共卫生和伦理而言都是双赢。

核心目标
通过采用科学验证的新方法论(例如如器官芯片、计算模型、体外检测等),逐步减少临床前安全研究中对动物实验的依赖,提升药物安全评估的预测准确性,同时降低研发成本和时间。
关键背景
1、动物实验的局限性:
-
90%通过动物实验的药物因安全性/有效性失败;
-
物种间生理差异导致误判(例如TGN1412单抗在动物中安全但引发人体严重免疫反应);
-
伦理争议及高昂成本(单抗开发需上百只灵长类动物,开发总成本超 7.5 亿美元)。
2、政策支持:
-
《FDA现代化法案2.0》(2022)允许非动物替代方法用于 IND 申请;
-
FDA科学委员会(2024)建议加速新方法论的验证。
3、技术进展:
-
器官芯片(例如肝脏芯片预测药物肝损伤的准确率高达 87%)、AI 模型、类器官等技术日趋成熟。
实施策略
1、短期(3年内)行动:
-
聚焦单克隆抗体(mAb):减少灵长类动物长期毒性试验(从6个月缩短至3个月);
-
数据整合:建立国际药物毒性数据库,利用现有临床数据替代部分动物实验;
-
试点项目:允许制药企业提交新方法论数据替代动物实验。
2、技术路径:
-
体外系统:多器官芯片模拟人体反应,免疫类器官检测细胞因子释放;
-
计算机模型:AI 预测抗体免疫原性、PBPK 模型优化药代动力学;
验证流程:通过回顾性分析(对比历史数据)和前瞻性试验验证新方法论可靠性。
3、跨部门协作:
-
联合国立卫生研究院(NIH)、退伍军人事务部(VA)等机构共享数据和资源,加速新方法论标准化与验证;
-
建立联邦级毒理学数据库(CAMERA),推动国际监管协调。
政策与激励
1、监管调整:
-
修订 ICH 指南,明确新方法论替代动物实验的条件;
-
发布案例指南,展示新方法论在IND/BLA中的成功应用。
2、企业激励:
-
对使用新方法论的企业提供快速审批通道;
-
公开“无动物实验获批药物”案例,树立行业标杆。
3、长期愿景(3-5年):
-
动物实验成为“例外”,仅用于新方法论无法解决的特定问题;
-
全球监管框架统一,推动新方法论成为药物安全评估新标准。
预期效益
科学:提升人体相关性,减少临床失败风险;
经济:缩短药物研发周期(当前平均研发周期为 9 年),降低单抗开发成本(当前平均开发成本为 7 亿美元);
伦理:减少非人灵长类等动物的使用。
FDA 通过整合类器官/器官芯片、AI 计算模型、体外检测等新方法论技术以及政策改革和跨部门协作,旨在建立以人体为中心的药物安全评估体系,推动监管科学进入“后动物实验时代”。这一路线图不仅提升公共健康保障,也有助于巩固美国在生物医药创新中的全球领导地位。
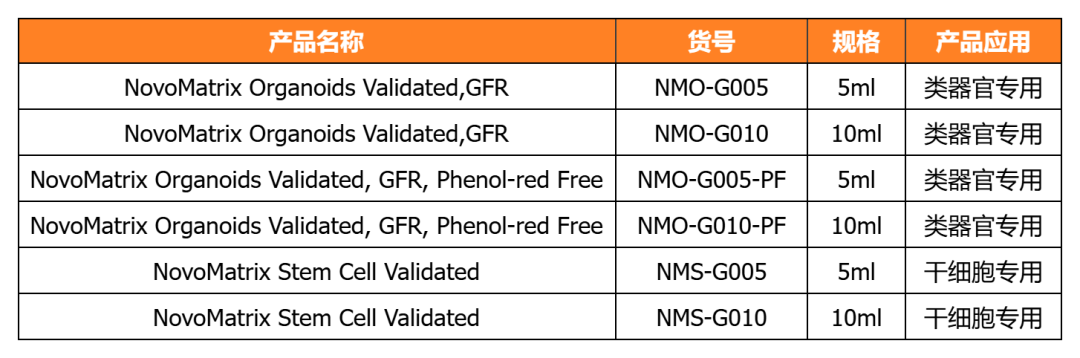
近岸蛋白相关产品
近岸蛋白提供多种类器官体外培养全方案,包括基质胶、培养基、相关细胞因子等,助力类器官研发国产化替代:
经类器官培养验证的完全培养基
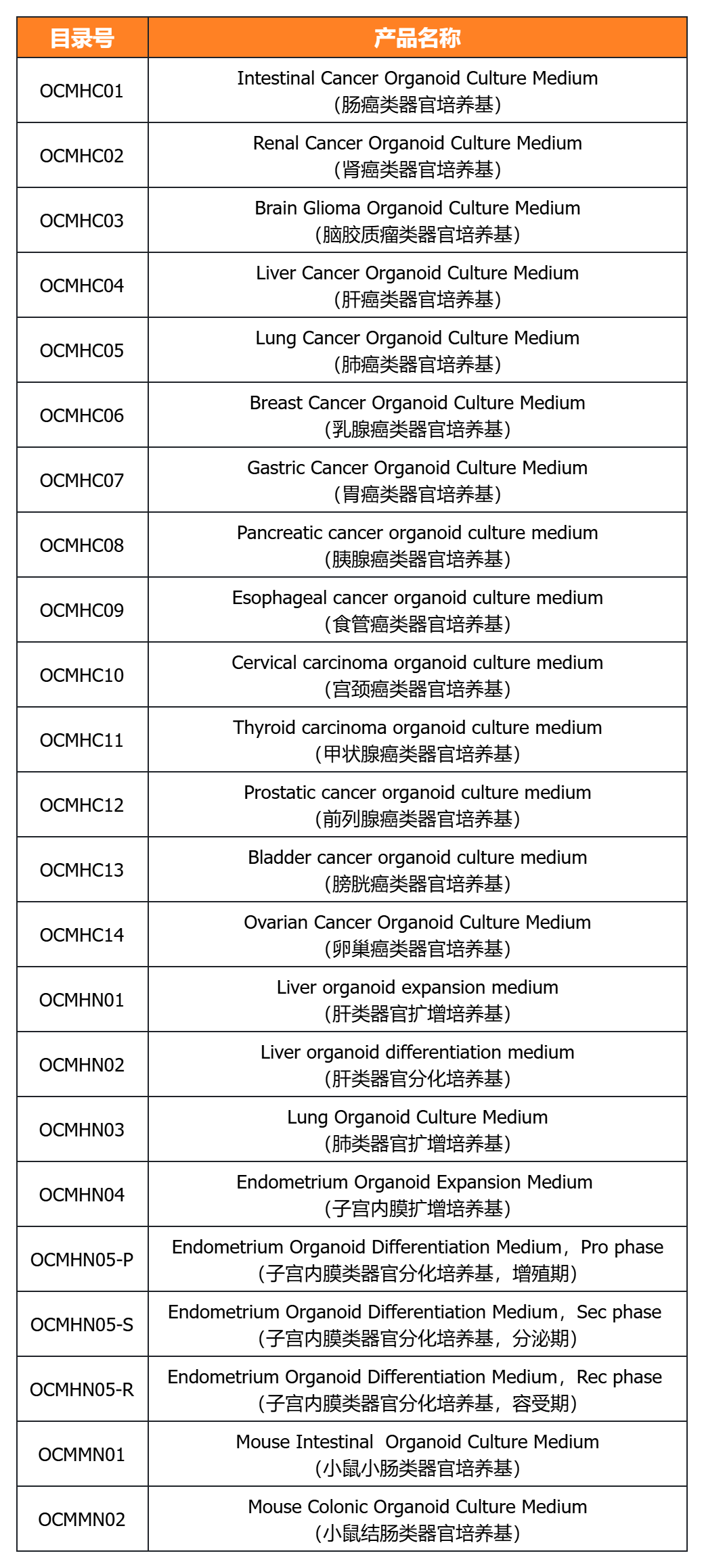
经类器官培养验证的细胞因子
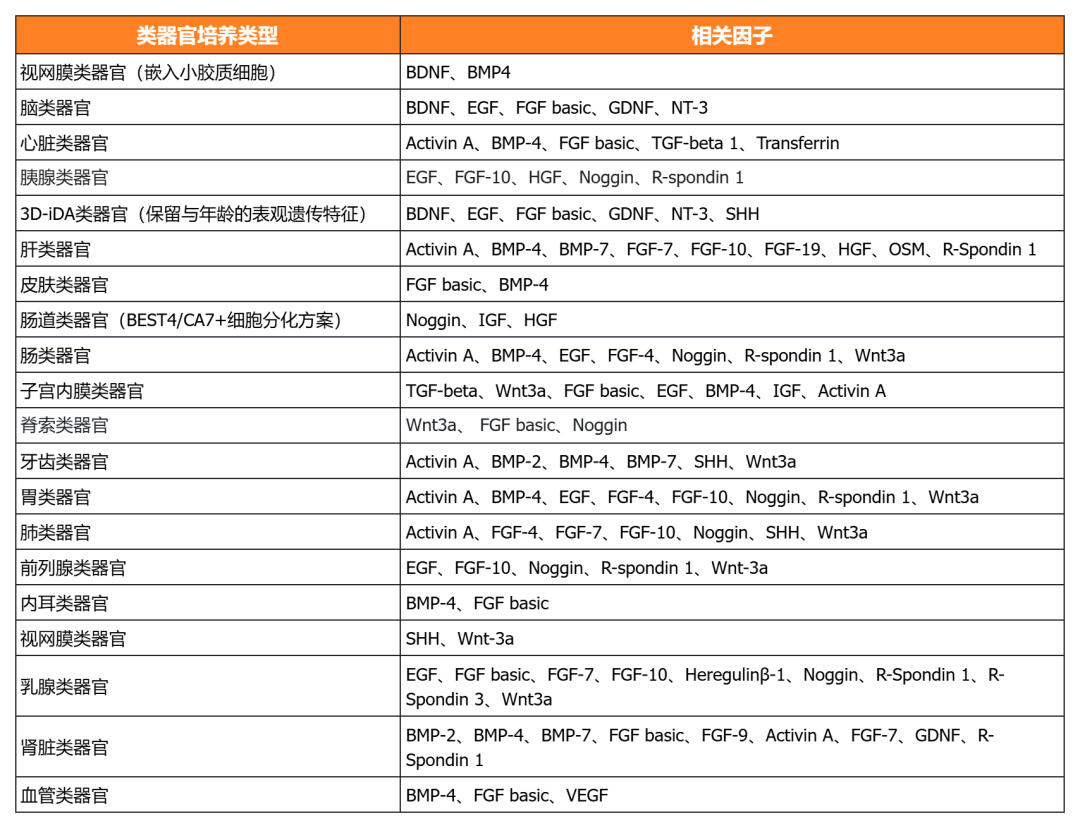
NovoMatrix 基质胶