
英文标题:Metabolic signaling of ceramides through the FPR2 receptor inhibits adipocyte thermogenesis
中文标题:神经酰胺通过FPR2介导的代谢信号传导抑制脂肪细胞产热
发表期刊:Science
影响因子:44.7
研究背景
神经酰胺是鞘磷脂代谢关键产物,与多种代谢疾病密切相关,但其作为系统信号分子的体内作用机制尚不明晰。脂肪细胞产热对维持机体能量平衡和体温稳态至关重要,cAMP信号通路参与其中,G蛋白偶联受体(GPCRs)在调控脂肪细胞产热中起核心作用。目前,虽已深入研究GPCRs对脂肪细胞产热的调控,但神经酰胺与GPCRs的交互关系及对脂肪细胞产热的调节作用研究较少。本研究聚焦于此,期望揭示相关调控机制,为解析神经酰胺在代谢疾病中的作用机制提供新思路,助力开发代谢疾病创新治疗策略。
研究成果
1、C16:0神经酰胺对脂肪代谢相关功能及FPR2受体信号通路的影响
研究者以C16:0神经酰胺为例,研究神经酰胺是否直接与GPCRs相互作用,启动跨膜信号传导,调节棕色和米色脂肪细胞的产热功能(图1A)。给正常饮食(NCD)喂养的野生型(WT)小鼠腹腔注射溶剂或C16:0神经酰胺,持续3天。在冷暴露期间,给予外源性C16:0神经酰胺会降低小鼠的耗氧量和能量消耗,但不影响其食物摄入量和运动活性(图1B-C)。
细胞实验方面,研究者使用C16:0神经酰胺对棕色脂肪组织(BAT)、皮下白色脂肪组织(scWAT)以及成熟脂肪细胞进行短暂刺激(刺激时长为15分钟)时,这些组织和细胞内的cAMP含量呈现出明显的下降趋势(图1D),这提示Gi偶联受体可能参与了神经酰胺对脂肪组织的急性作用。
为进一步探究,研究者借助GSE40486的微阵列数据,筛选出小鼠棕色脂肪组织中高表达的60种GPCRs。后续利用GloSensor-cAMP检测法对这些受体展开筛选,发现在过表达FPR2的人胚肾293(HEK293)细胞中加入C16:0神经酰胺后,由佛司可林诱导产生的cAMP水平呈剂量依赖性下降(图1E)。使用Gαi1和Gαi2解离实验进一步验证,结果表明人源hFPR2和小鼠源mFPR2这两种不同物种的GPCRs对其内源配体的反应有时会有所不同(图1F)。具体而言,C16:0神经酰胺在Gi信号通路中激活hFPR2或mFPR2的效能与其他已知的内源性配体(如fMLFII、Ac2-26、LL-37、humanin)相当,但功效较低,可能是FPR2的部分激动剂。
研究者运用G蛋白解离和β-arrestin1募集实验,探索原代脂肪细胞内源性FPR2的响应。构建含G蛋白及β-arrestin1 BRET探针的慢病毒感染原代脂肪细胞,并以Adipoq-Cre+/−Fpr2fl/fl小鼠作阴性对照。结果显示,从Fpr2fl/fl小鼠分离的原代脂肪细胞,其Gi信号通路可被激活,且对C16:0神经酰胺、LL-37和fMLFII呈浓度依赖性反应,而阴性对照的原代脂肪细胞无此反应(图1G)。
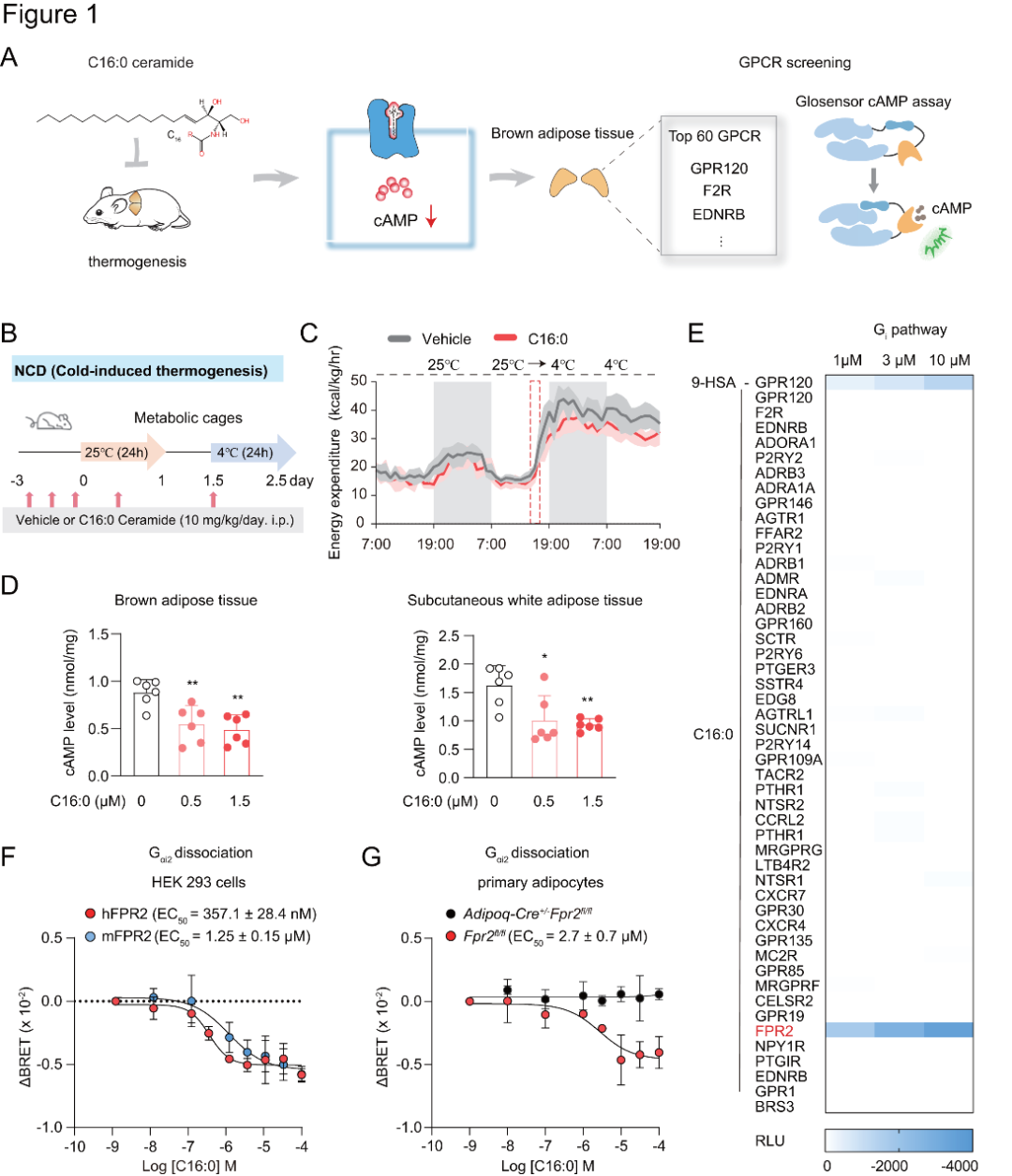
图1.神经酰胺膜受体的鉴定
2、FPR2与神经酰胺的直接相互作用
研究者使用FPR2抗体,研究了野生型小鼠的scWAT和BAT中FPR2的表达模式,并以Fpr2基因缺陷型(Sox2-CreER+/−Fpr2fl/fl)小鼠作为阴性对照。结果表明,FPR2主要分布在野生型小鼠的scWAT或BAT中表达脂联素的脂肪细胞内,而在Fpr2基因缺陷型小鼠中则不存在这种分布(图2A-B)。
为探究C16:0神经酰胺与FPR2的直接结合特性,研究者采用两种方法,(1)体外实验:在竞争性结合实验中,用FITC标记的高亲和力肽激动剂WK(FITC)YMVm作用于FPR2。构建含Nanoluc荧光基序且与hFPR2的N端融合的质粒,在HEK293细胞中表达,检测WK(FITC)YMVm的结合情况。发现C16:0神经酰胺浓度增加时,WK(FITC)YMVm与Nluc-FPR2间的BRET信号减弱;(2)体内实验:合成125I标记的FPR2肽激动剂125I-Ac2-26。饱和结合分析显示,其能特异性结合野生型小鼠BAT、scWAT及过表达hFPR2的HEK293细胞的膜组分。竞争性结合实验证实,C16:0神经酰胺可与125I-Ac2-26竞争结合上述膜组分(图2C)。
已知GPCR结合会使细胞外结构域发生选择性构象变化,为探究FPR2是否如此,研究者构建FlAsH BRET传感器,监测其细胞外环(ECL)区域在结合神经酰胺和fMLFII时的构象变化(图2D)。结果显示,C16:0神经酰胺使FPR2的N端靠近ECL1和ECL2区域,而fMLFII则使FPR2的N端远离ECL2区域(图2E)。这表明C16:0神经酰胺与FPR2结合会引发受体特定细胞外构象变化,可能影响其信号传导特异性。
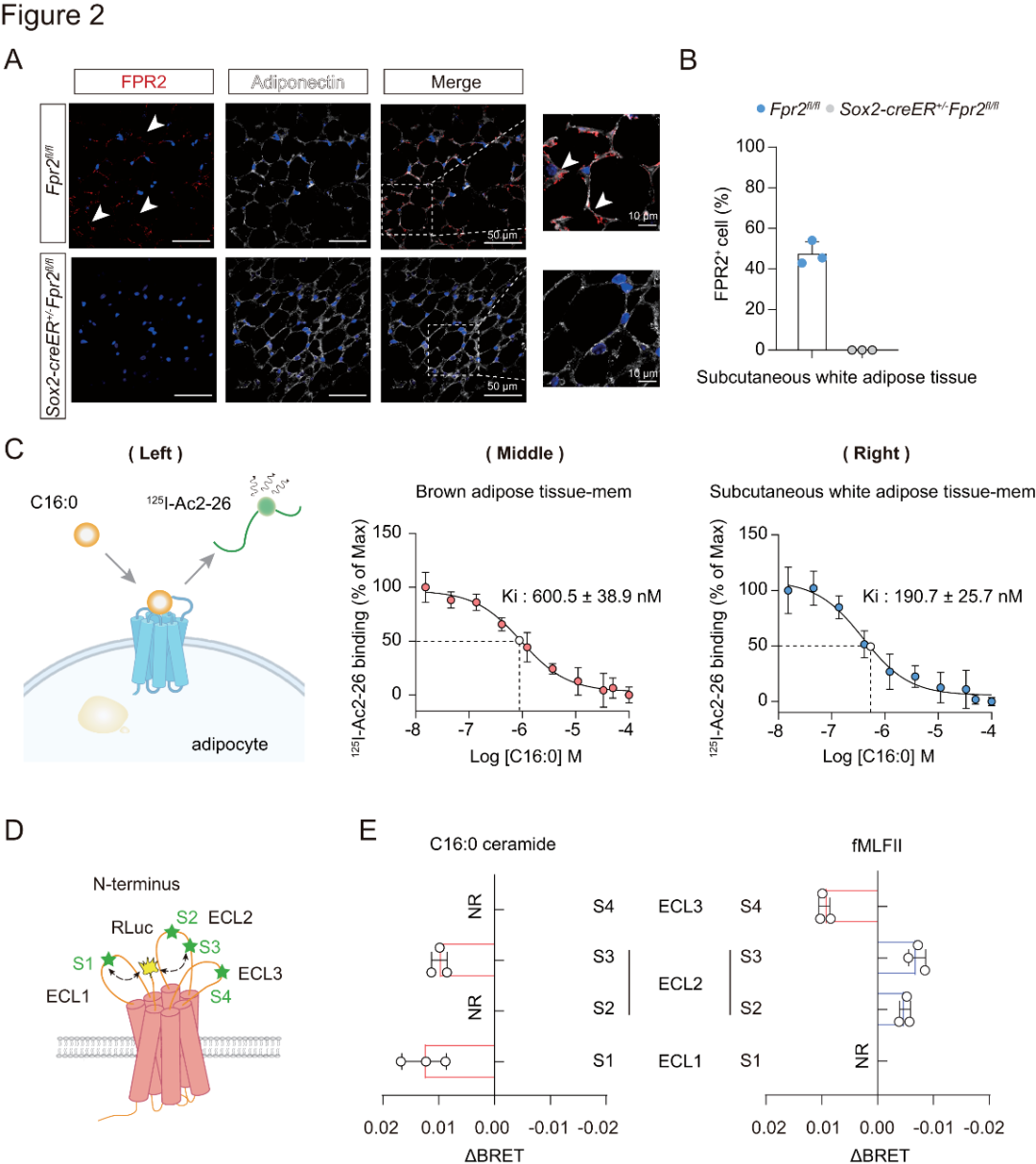
图2.脂肪细胞中C16:0神经酰胺与FPR2的直接相互作用
3、FPR2介导C16:0神经酰胺对产热的抑制作用
为探究C16:0神经酰胺对冷诱导产热的抑制是否依赖脂肪细胞FPR2表达,研究者构建了Ucp1-Cre+/-Fpr2fl/fl小鼠及Adipoq-Cre+/-Fpr2fl/fl小鼠。对两类基因编辑小鼠及对照Fpr2fl/fl小鼠,喂正常饮食,腹腔注射溶剂或C16:0神经酰胺(图3A)。结果显示,C16:0神经酰胺可抑制Fpr2fl/fl小鼠在室温和寒冷下的能量消耗与耗氧量,但在Adipoq-Cre+/-Fpr2fl/fl及Ucp1-Cre+/-Fpr2fl/fl小鼠中无此效果(图3B-C)。
研究者进一步研究了冷暴露后C16:0神经酰胺通过FPR2对脂肪细胞产热的影响。给喂食正常饮食的Adipoq-Cre+/-Fpr2fl/fl小鼠、Ucp1-Cre+/-Fpr2fl/fl小鼠以及对照Fpr2fl/fl小鼠腹腔注射溶剂或C16:0神经酰胺。为探究C16:0神经酰胺对BAT活性和米色脂肪生成的抑制作用是否依赖于FPR2,在冷暴露一天后检测了小鼠BAT和scWAT中产热相关基因的相对表达(图3D)。C16:0神经酰胺处理降低了对照组小鼠的直肠温度以及腹股沟和肩胛间区域的局部温度,同时降低了UCP-1蛋白水平和产热基因的表达(图3E-F)。H&E染色和免疫组化进一步证实,C16:0神经酰胺处理抑制了Fpr2fl/fl小鼠的BAT活性和米色脂肪生成(图3G)。然而,在两类基因编辑小鼠中,C16:0神经酰胺的上述作用消失了(图3E-G)。白色脂肪组织中Fpr2基因缺乏可上调UCP-1等产热相关基因表达,且不依赖外源性C16:0神经酰胺(图3D-G)。综上,在生理条件下,内源性神经酰胺可能通过激活FPR2抑制白色脂肪组织向棕色脂肪的转化及产热过程(图3呈现了Adipoq-Cre+/-Fpr2fl/fl小鼠在相关实验中的结果,Ucp1-Cre+/-Fpr2fl/fl小鼠的实验结果详情可查阅原文的补充材料)。

图3.C16:0神经酰胺通过FPR2抑制冷暴露诱导的产热作用
4、FPR2与神经酰胺复合物的整体结构及识别特异性
神经酰胺的脂肪链长度以及特定位置上不饱和的碳碳双键存在差异,会影响其生理功能。因此,研究者探究了各种脂质结构对FPR2的作用活性(图4A-B)。神经酰胺的亚结构,如棕榈酸(PA)、花生酸(AA)和鞘氨醇,不能激活FPR2下游的Gi信号通路(图4B)。只有具有饱和脂肪链(C2:0、C6:0、C10:0、C16:0、C18:0和C20:0)的神经酰胺才能激活FPR2(图4B)。带有不饱和碳碳双键(C18:1和C24:1)的神经酰胺以及超长链神经酰胺无法激活FPR2(图4B)。
为了剖析饱和长链神经酰胺是如何被膜受体FPR2特异性识别的,研究者使用单颗粒冷冻电子显微镜(cryo-EM)来确定与神经酰胺和Gi1形成复合物的人类FPR2的结构,重构、纯化了C16:0、C18:0和C20:0神经酰胺-FPR2-Gi1-scFv16复合物(图4C)。神经酰胺呈 “p” 形垂直插入FPR2正构结合位点,与fMLFII和humanin结合方式相似,区别于与Gq偶联的神经酰胺受体CYSLTR2(图4D)。
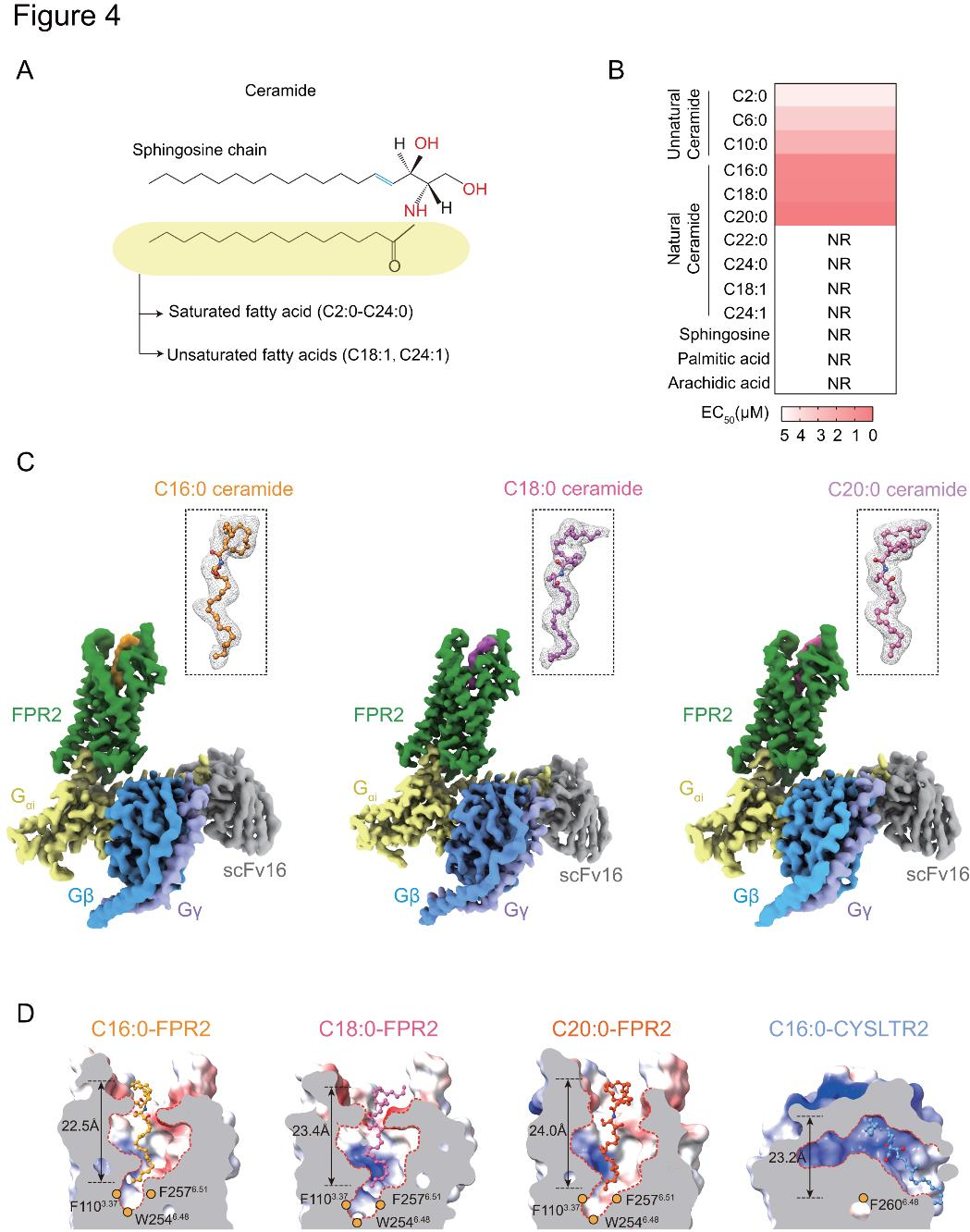
图4.hFPR2-Gi配合物与神经酰胺结合的整体结构
5、神经酰胺在FPR2正构位点的结合
在C16:0、C18:0和C20:0神经酰胺-FPR2-Gi1复合物结构中,神经酰胺由C18鞘氨醇链与N-酰基链经中心酰胺键连接,其鞘氨醇脂肪链深入FPR2配体口袋,与特定芳香残基作用并占据fMLFII位置(图4D)。脂肪链位于由L109³.³⁶、F110³.³⁷、R201⁵.³⁸、R205⁵.⁴²和F257⁶.⁵¹组成的口袋中(图5A-D)。L109³.³⁶A、F110³.³⁷A、R201⁵.³⁸A、R205⁵.⁴²A和F257⁶.⁵¹A的突变会降低神经酰胺诱导的FPR2激活(图5F)。特别是,H102³.²⁹和F178ECL2可能与神经酰胺的不饱和C=C双键形成长距离的π-π堆积(图5A-D)。H102³.²⁹L/A或 F178ECL2L/A的突变显著降低了这些神经酰胺诱导的FPR2激活(图5F)。脂肪酸部分脂肪链折叠成特定结构,围绕其疏水口袋残基突变会损害FPR2对神经酰胺刺激的激活,表明FPR2细胞外疏水环境有助于结合(图5F)。由于FPR2的配体口袋较大,且神经酰胺的脂肪链具有柔性,研究者进行了分子动力学模拟,以研究在冷冻电镜结构中未观察到的潜在相互作用。发现E89ECL1等残基与神经酰胺中心羧化鞘氨醇基团相互作用(图5E)。这些残基的丙氨酸突变降低了FPR2对神经酰胺刺激的激活(图5F)。
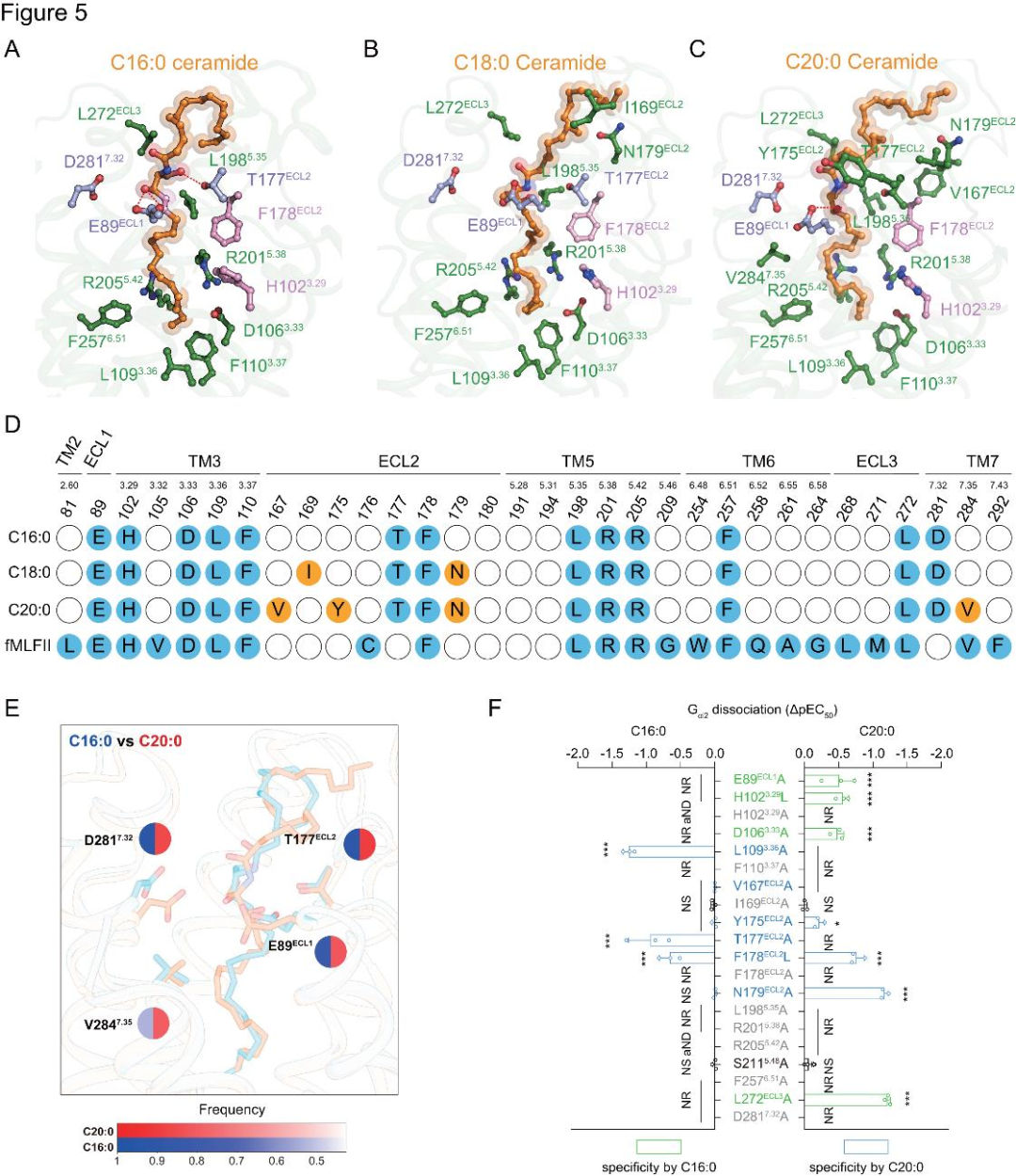
图5.FPR2对神经酰胺的识别
6、FPR2对神经酰胺识别特异性的调控机制
在C16:0(p构象)、C18:0(s构象)及C20:0(s构象)神经酰胺与FPR2相关复合物结构中,神经酰胺脂肪酸部分呈弯曲构型(图4C和图5A-C),而脂肪酸链中C=C双键的存在可能会破坏此构型,致不饱和神经酰胺无法与FPR2稳定互作(图6C-D)。饱和Cn:0神经酰胺活性随脂肪酸碳链增长而增强,达22个碳原子时活性消失(图4A-B)。在相关复合物中,神经酰胺脂肪酸脂肪链被跨膜结构域5(TM5)、ECL2和ECL3细胞外疏水残基包围(图6A),碳链增长可增加与FPR2疏水作用(图6B)。这些残基突变对C18:0、C20:0诱导的FPR2激活影响大于C16:0,表明FPR2区域疏水口袋可塑性对容纳长链神经酰胺很重要,且计算模拟显示该口袋对极长链神经酰胺而言可能过小(图6E)。
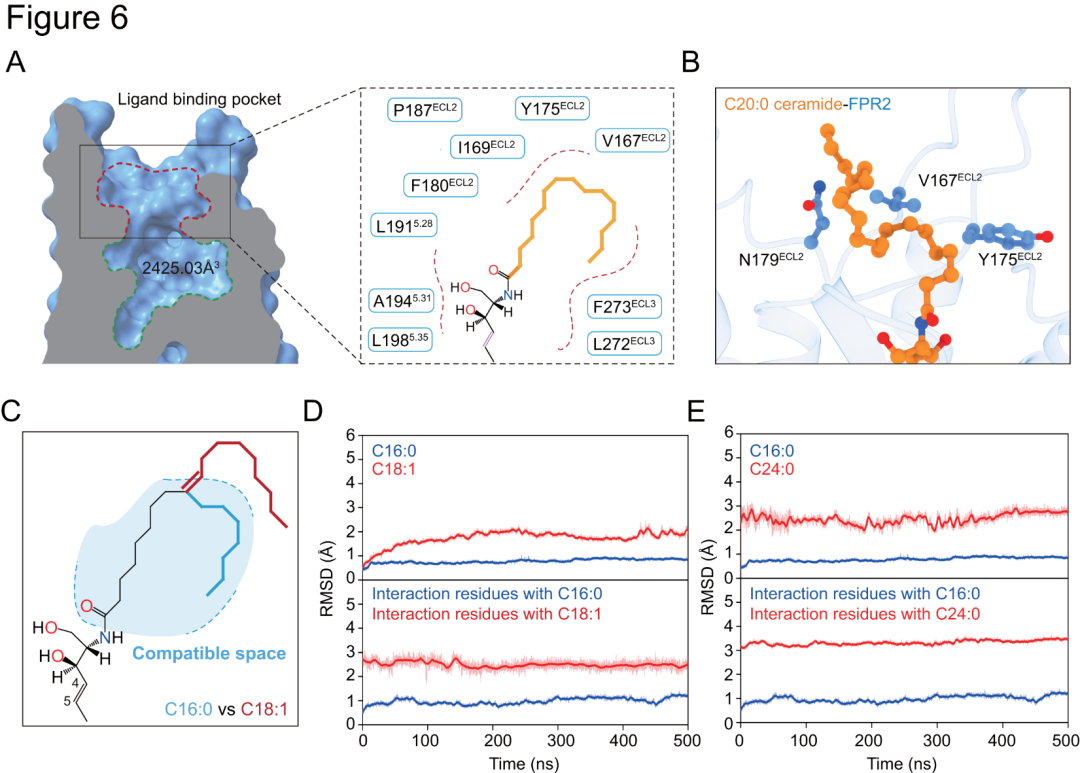
图6.FPR2对不同神经酰胺选择性识别的结构基础
7、神经酰胺对FPR2及其相关受体的选择性作用
为探究神经酰胺信号传导特异性,研究者测试了C16:0神经酰胺对6种与FPR2密切相关的GPCR(FPR3、FPR1、GPR32、C5R1、CMKLR1和GPR1)的活性影响。结果显示,这些受体对C16:0神经酰胺均无响应(图7A-B)。其中,FPR1和FPR3与FPR2亲缘关系最近(图7C),但FPR2中识别羧化鞘氨醇基团或不饱和C=C的关键残基,如E89ECL1、D281⁷.³²和H102³.²⁹,在FPR1中被替换(图7C-D),相应突变降低了神经酰胺对FPR2的激活,致使神经酰胺无法激活FPR1(图7F)。FPR3中L198⁵.³⁵、R201⁵.³⁸和R205⁵.⁴²发生替换(图7C和E),FPR2中对应突变也降低C16:0神经酰胺的激活作用(图7F)。除FPR成员外,其他进化上接近FPR2的受体无“EECL1&H³.²⁹”基序,FPR2相应突变损害C16:0神经酰胺诱导的Gi活性(图7C和F)。相反,FPR1的G89ECL1E突变和 FPR3的A198⁵.³⁵L-H205⁵.⁴²R组合突变,使这两种受体能够感知C16:0神经酰胺(图7G-J)。以上结果确定E89ECL1、L198⁵.³⁵和R205⁵.⁴²为识别神经酰胺的关键决定因素。
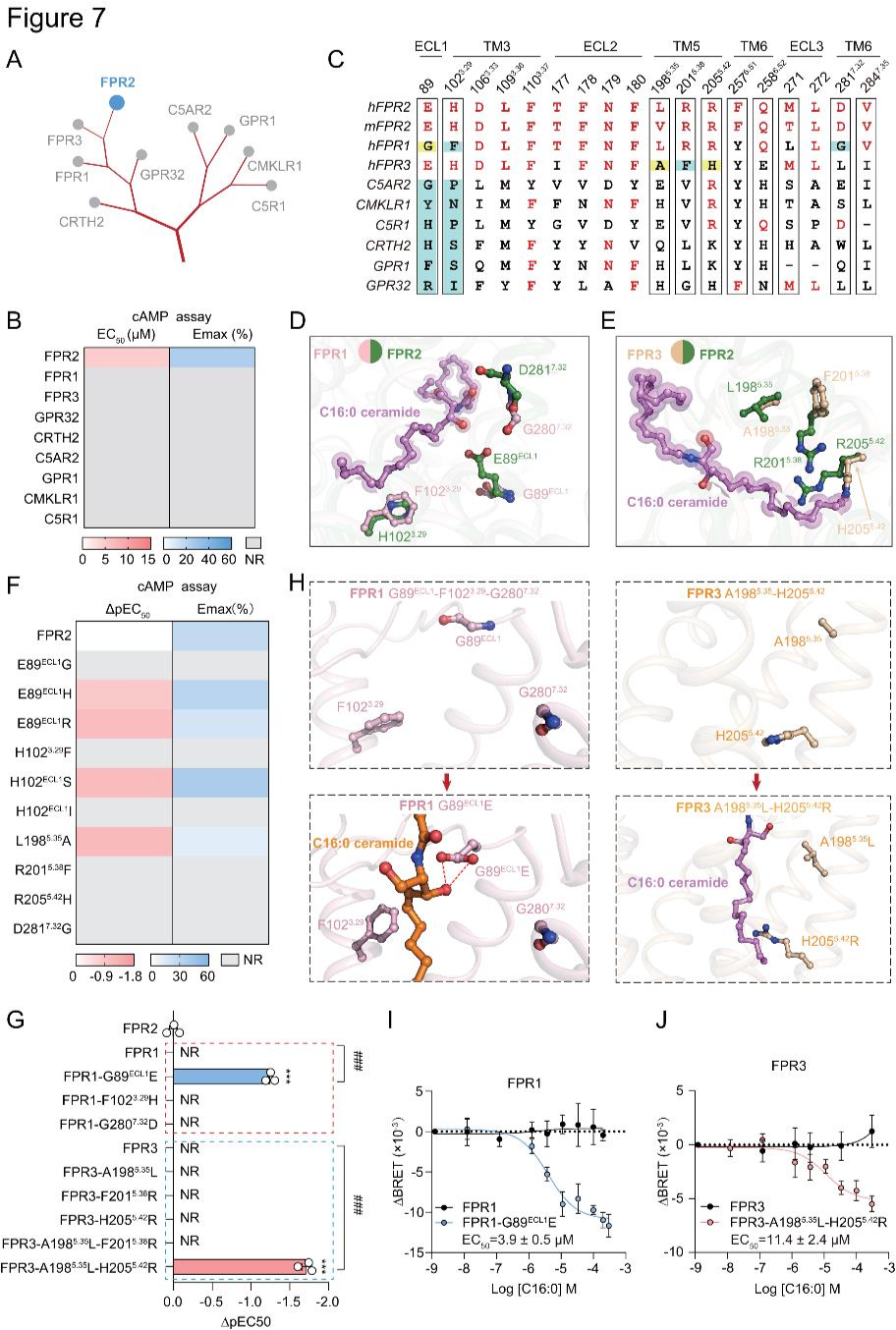
图7.FPR2是C16:0神经酰胺的特异性受体
8、神经酰胺激活FPR2的潜在机制
为了探究神经酰胺激活FPR2的机制,研究者基于AlphaFold蛋白质结构数据库预测结构,在脂双层中进行分子动力学松弛处理后,构建了无配体FPR2模型(图8A-B)。通过对比神经酰胺-FPR2-Gi复合物的冷冻电镜结构与该模型,发现关键构象变化(图8C-G)。C16:0神经酰胺的鞘氨醇脂肪链与特定残基相互作用,引发残基旋转、移动,促使作为转换开关的W254⁶.⁴⁸与保守基序堆积,使神经酰胺结合诱导的构象变化经转换开关传至相关基序及细胞质区域(图8E)。这一过程中,无配体FPR2结构中的特定盐桥和氢键被破坏,新的极性相互作用网络形成(图8F)。同时,C16:0-FPR2-Gi结构中,FPR2的TM7向外移动,引发相关基序重排,特定残基间形成额外的π-π堆积和π-阳离子堆积(图8G)。
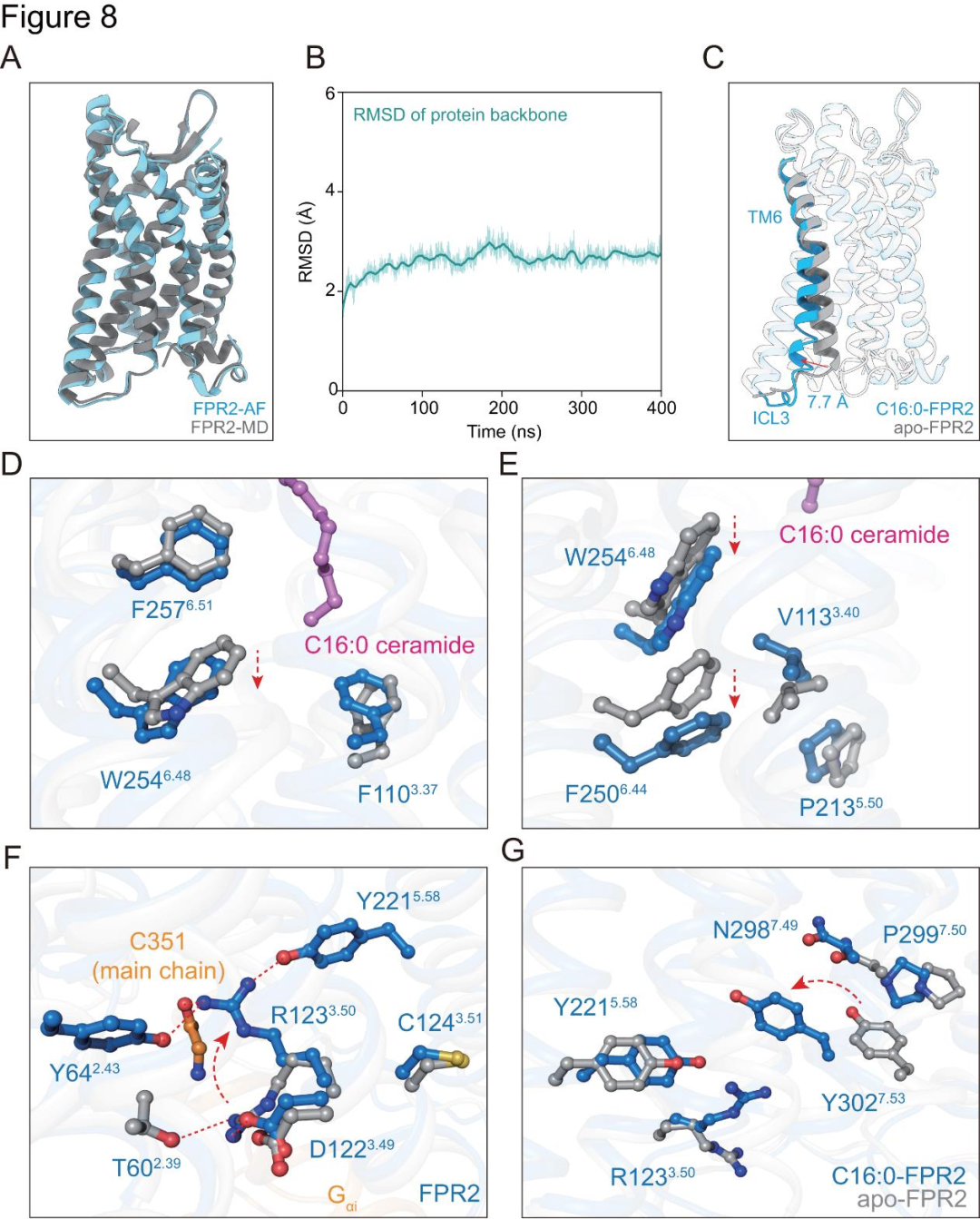
图8.FPR2与C16:0神经酰胺结合的激活机制
研究小结
神经酰胺作为鞘磷脂代谢核心分子,其异常累积与糖尿病、肥胖及动脉粥样硬化等代谢疾病密切相关。近期研究发现,C16:0神经酰胺可以直接与脂肪细胞中的神经酰胺膜受体FPR2结合,激活Gi信号通路,降低细胞内cAMP水平,进而抑制UCP-1表达,降低产热能力,基因敲除或拮抗剂实验已证实FPR2的介导作用。研究团队利用冷冻电镜解析神经酰胺-FPR2-Gi复合物结构,揭示FPR2对神经酰胺的选择性识别机制。该研究成果为深入剖析神经酰胺的生理功能打开了全新的窗口,为代谢性疾病靶向治疗开拓了新方向。
END
Peng 撰文
Orianna 校稿